Menu

In the era of precision oncology, it has become increasingly common for patients diagnosed with cancer to undergo tumor sequencing. Identifying the mutations that make up a tumor’s genomic landscape can help guide selection of targeted therapies and inform prognosis. Despite the recognized value of tumor-only sequencing, labs performing this type of testing face a number of technical challenges that, if not properly addressed, can render the results uninformative or even misleading.
Although there are a variety of inherent challenges in tumor-only sequencing, all ultimately impact the ability to accurately distinguish somatic mutations driving tumorigenesis from germline variants associated with cancer predisposition. In fact, it has been estimated that as many as one third of mutations identified by tumor-only sequencing may be false-positive germline changes, including in potentially actionable genes1. Having an accurate picture of a tumor’s genomic makeup and contextual genetic environment is crucial to an accurate clinical assessment, which impacts therapeutic recommendations and represents the patient’s best chance for successful treatment.
In this blog we explore different strategies for enriching tumor analysis for somatic mutations and discuss why matched tumor-normal sequencing has become the preferred method.
One approach is to use variants present in large population databases as a filter to remove likely germline variants from a tumor sample2. While this practice is generally effective, it will also remove true somatic variants that happen to be identical to germline variants, resulting in a false negative. Database-driven approaches can also overlook any rare germline variants missing from large population databases due to underrepresentation of non-White individuals. These variants will remain in the sequencing data and can result in false-positive germline findings.
Taking allele frequency into consideration can help. This strategy is based on the premise that an allele frequency of 50% is consistent with a heterozygous germline variant, and an allele frequency of ~100% is consistent with a homozygous germline variant1. It then stands to reason that focusing on variants with a lower allele frequency increases the likelihood of somatic origin.
While this is true, such an approach can be complicated by many factors including contamination of the tumor sample with normal tissue, tumor heterogeneity, sequencing artifacts, difficulty mapping reads in regions of high homology, high level mosaic variants that arose early in differentiation, as well as changes in allele fraction due to copy number changes or loss of heterozygosity (LOH). Any of which can lead to inaccurate attribution of origin.
Matched tumor-normal sequencing that pairs analysis of a tumor sample with that of a comparable, normal sample – most often from the same individual – has been shown to be a more effective strategy, yielding more reliable identification of the somatic changes specific to a tumor1,3,4. As the name suggests, variants in the matched normal sample are determined to be germline in origin, or of alternate origin unrelated to the current tumorigenicity. When used as a filter against the tumor sample, somatic variants relevant to the cancer at hand can be identified with a high degree of confidence. Variants found at low frequencies in the normal sample can be confidently classified as false positives if they are not significantly enriched in the tumor.
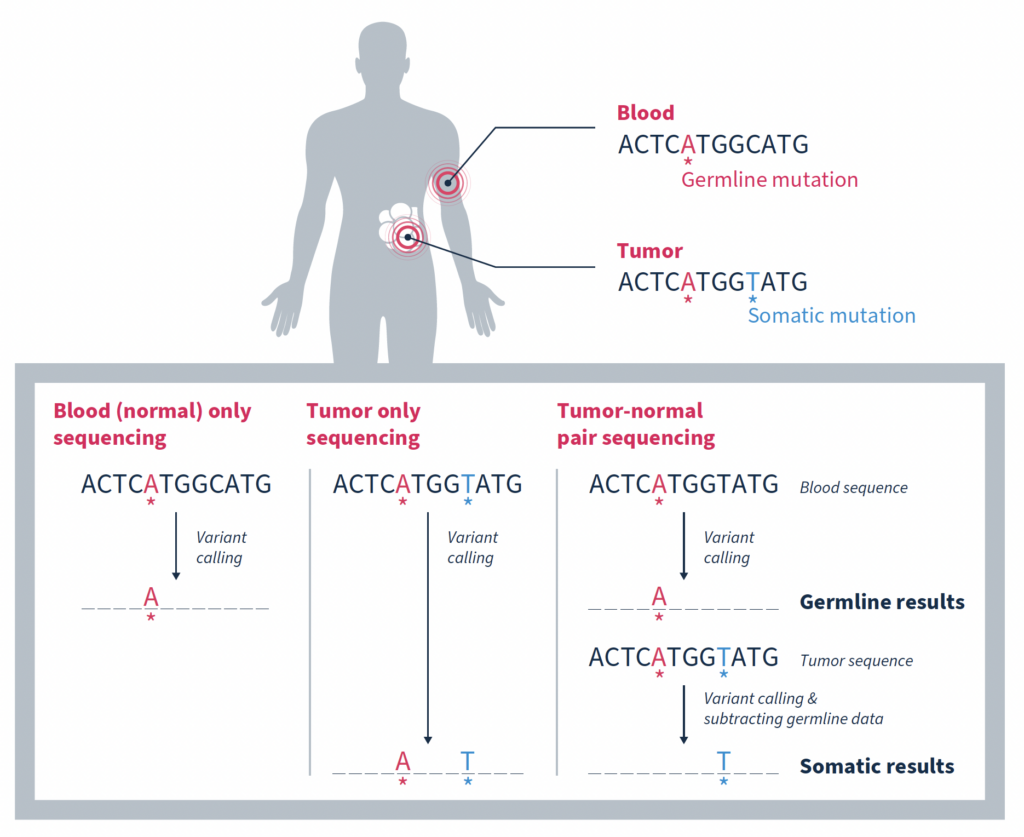
While the most important function of matched tumor-normal sequencing is to identify and retain somatic mutations, it also serves other important functions.
At the most simplistic level, biological samples can exhibit variability due to factors such as environmental influences, biological processes and sample handling. Matched-tumor normal sequencing provides a built-in baseline of background noise resulting from these factors, or from introduction of sequencing artifacts, that can be filtered out.
In the case of FFPE samples, extracted DNA is often fragmented and of a lower quality than fresh tissue samples. Matched tumor-normal sequencing provides a comparison that helps distinguish true alterations from noise resulting from degradation of the DNA, enhancing sensitivity.
Cell-free DNA (cfDNA) samples, also known as liquid biopsy samples, contain DNA from tumor cells, but they also contain a significant amount of DNA from white blood cells. In many individuals, especially those who are older, these phenotypically normal blood cells contain acquired mutations subsequently increased in relative frequency due to clonal expansion. These clonal hematopoiesis of indeterminate potential (CHIP) variants often, but not always, occur in the same genes associated with blood cancers like leukemia. However, while they are indicative of an increased risk of developing a blood cancer in the future, they are not likely to be relevant to the tumor being analyzed.
Simultaneously sequencing matched white blood cells as a normal control can successfully distinguish somatic mutations that are relevant to driving tumorigenesis from somatic mutations arising from the normal process of clonal hematopoiesis4. This is such an important consideration that both ESMO and AMP guidelines specify that matched white blood cell sequencing should be used for interpretation of somatic variants in cfDNA testing5,6.
Removal of false positives arising from CHIP variants is not only important for accurate cfDNA analysis, but also FFPE analysis. In a study by Memorial Sloan Kettering Cancer Center (MSK) investigators, matched tumor-normal sequencing results showed that 5.2% (912/17,469) of patients with advanced cancer would have had at least 1 clonal hematopoietic (CH)-associated mutation erroneously called as tumor-derived in the absence of matched blood sequencing7. Of these CH variants, 49.7% of them were classified as oncogenic or likely oncogenic based on OncoKB™, and 3.2% were associated with approved or investigational therapies (e.g. mutations in IDH1/2). Failure to recognize such mutations as blood-derived may result in inaccurate precision therapy recommendations.
The ability to distinguish between somatic and germline variants has the additional benefit of streamlining analysis of germline variants which have additional implication for a patient’s clinical care. Notably providing information about future disease risk which can be managed in part through surveillance as well as allowing for testing of family members who may also be at risk for disease.
It is for the reasons discussed here that MSK-ACCESS® powered with SOPHiA DDM™ for liquid biopsy and MSK-IMPACT® powered with SOPHiA DDM™ for comprehensive genomic profiling (CGP) utilize the matched tumor-normal analysis strategy to accurately delineate somatic variants from germline and CHIP variants.
Contact us to learn more about adopting advanced liquid biopsy and CGP technology in your laboratory.
References
SOPHiA GENETICS products are for Research Use Only and not for use in diagnostic procedures unless specified otherwise.
SOPHiA DDM™ Dx Hereditary Cancer Solution, SOPHiA DDM™ Dx RNAtarget Oncology Solution and SOPHiA DDM™ Dx Homologous Recombination Deficiency Solution are available as CE-IVD products for In Vitro Diagnostic Use in the European Economic Area (EEA), the United Kingdom and Switzerland. SOPHiA DDM™ Dx Myeloid Solution and SOPHiA DDM™ Dx Solid Tumor Solution are available as CE-IVD products for In Vitro Diagnostic Use in the EEA, the United Kingdom, Switzerland, and Israel. Information about products that may or may not be available in different countries and if applicable, may or may not have received approval or market clearance by a governmental regulatory body for different indications for use. Please contact us at [email protected] to obtain the appropriate product information for your country of residence.
All third-party trademarks listed by SOPHiA GENETICS remain the property of their respective owners. Unless specifically identified as such, SOPHiA GENETICS’ use of third-party trademarks does not indicate any relationship, sponsorship, or endorsement between SOPHiA GENETICS and the owners of these trademarks. Any references by SOPHiA GENETICS to third-party trademarks is to identify the corresponding third-party goods and/or services and shall be considered nominative fair use under the trademark law.


