Menu


The transition from the In Vitro Diagnostic Directive (IVDD) to the In Vitro Diagnostic Regulation (IVDR) in the European Union marks an important advancement in regulatory standards for genetic testing and analysis. The new standards promote transparency and traceability throughout genomic analysis processes, helping to ensure the reliability and accuracy of diagnostic results and ultimately patient safety.
Everyone wants to ensure that genomic analysis is safe for patients. However, healthcare institutions face real challenges in transitioning from IVDD to IVDR. Particularly with the use of complex software solutions for genomic data analysis, many of which are designated as research use only (RUO).
One of the biggest changes for healthcare institutions is that IVDR specifically regulates in-house manufactured tests. With few exceptions, healthcare institutions utilizing in-house manufactured tests must now meet the same requirements and proof of conformity with IVDR as manufacturers. These requirements extend to the software that is used for the analysis, interpretation and reporting of NGS data. Software developed in-house, including from public domain materials, must meet many of the same requirements and proof of conformity as commercial software solutions.
Here we answer some of the most pressing questions about what IVDR compliance entails for healthcare institutions performing genomic analysis and how CE-IVD certified software solutions can help.
Q: How does IVDR impact the analysis, interpretation, and reporting of NGS data?
A: Software used to support the analysis, interpretation and reporting of NGS data from genetic testing must conform to IVDR’s general safety and performance requirements (GSPR) to ensure reliability and safety.
Q: What are the key requirements for meeting IVDR compliance?
A: Genetic tests and their analytical software require that a healthcare institution perform the following to ensure IVDR compliance:
It is also important to ensure and demonstrate that suppliers are complying with applicable regulatory requirements.
After launch, post-market surveillance is required to monitor the safety and clinical performance of the test. Genetic tests require regulatory management of updates as well as reporting of any serious incidents, with the corrective actions taken.
Q: When does IVDR take effect?
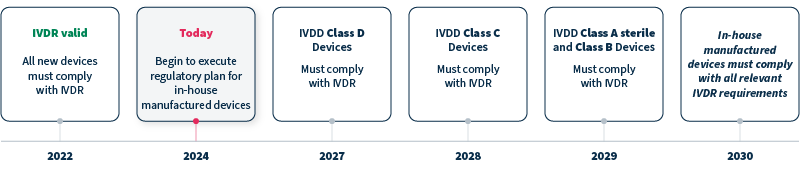
A: IVDR replaced IVDD in 2022, with timelines for compliance differing for devices with new, versus legacy, status and class. Genetic tests and analytical software that are Class C devices, for example, must comply with IVDR by 2028, and in-house manufactured devices must comply with all relevant IVDR requirements by 2030 at the latest (see timeline).
Q: How can commercial software solutions help with the IVDR transition?
A: When using a CE-IVD commercial software solution, the manufacturer’s CE-IVD certification and their existing technical documentation provide proof of compliance with current regulations, helping to lessen the burden for the healthcare institute.
Q: Can research use only (RUO) solutions be used to meet IVDR requirements?
A: RUO solutions are not intended or validated for clinical diagnostic use. RUO solutions will not be considered compliant without additional testing and validation as part of an in-house manufactured device.
Q: What are the most important considerations when evaluating commercial CE-IVD analytical software solutions?
A: When evaluating analytical software solutions, regulatory compliance is a must. However, it is also important to consider analytical and clinical validity. Does the solution provide reliable data processing and variant calling with high sensitivity and specificity? What variant types does it cover? Is it able to interpret variants according to established guidelines? Looking ahead to the future, can the solution scale with growing genomic analysis volumes?
As important as the new IVDR standards are to ensuring patient safety, it is clear that the transition poses a number of challenges to healthcare institutions performing genomic analysis. At SOPHiA GENETICS we’re proud to help simplify the transition, offering fast, reliable CE-IVD oncology applications powered by SOPHiA DDM™.
To learn more, explore our current portfolio and stay tuned for further updates.
SOPHiA GENETICS products are for Research Use Only and not for use in diagnostic procedures unless specified otherwise.
SOPHiA DDM™ Dx Hereditary Cancer Solution, SOPHiA DDM™ Dx RNAtarget Oncology Solution and SOPHiA DDM™ Dx Homologous Recombination Deficiency Solution are available as CE-IVD products for In Vitro Diagnostic Use in the European Economic Area (EEA), the United Kingdom and Switzerland. SOPHiA DDM™ Dx Myeloid Solution and SOPHiA DDM™ Dx Solid Tumor Solution are available as CE-IVD products for In Vitro Diagnostic Use in the EEA, the United Kingdom, Switzerland, and Israel. Information about products that may or may not be available in different countries and if applicable, may or may not have received approval or market clearance by a governmental regulatory body for different indications for use. Please contact us at [email protected] to obtain the appropriate product information for your country of residence.
All third-party trademarks listed by SOPHiA GENETICS remain the property of their respective owners. Unless specifically identified as such, SOPHiA GENETICS’ use of third-party trademarks does not indicate any relationship, sponsorship, or endorsement between SOPHiA GENETICS and the owners of these trademarks. Any references by SOPHiA GENETICS to third-party trademarks is to identify the corresponding third-party goods and/or services and shall be considered nominative fair use under the trademark law.


