More than six people die every hour in the US from a blood cancer. Solutions can’t come fast enough for those who suffer with these cancers all around the world. Fortunately, researchers studying blood diseases have experienced rapid advances in their capabilities to develop and test effective therapies with some extremely significant advancements.
1. Next-generation sequencing (NGS)
Some of the most difficult limitations of molecular profiling for hematological cancer disorders include accurate detection of mutations in GC-rich gene regions and insertions or deletions in challenging genes. Data analysis on NGS DNA samples identifies complex variants to accurately identify myeloid malignancies. This validation of targeted mutations has encouraged many medical centers to order NGS testing for every acute myeloid leukemia case.
Faster, more efficient NGS analysis can drive better hematological cancer research outcomes to potentially improve care for patients with blood cancers and diagnosis of new cases.
2. Guideline evolution
International guidelines for hematological cancer diagnosis and treatment are continuously evolving and create the need for laboratories’ fast adaptation. Those evidence-based guidelines by physician commissions contribute to improving the clinical standard of care. The World Health Organization, European Hematology Association, European LeukemiaNet, College of American Pathologists and the American Society of Hematology call for increased use of NGS testing for initial diagnostic workup of blood cancers.
Detection of the relevant biomarkers for myeloid malignancies by NGS, per international guidelines, helps to ensure optimal clinical trial enrollment, therapy validation, dose protocols and other research benefits. A solution that can be constantly updated and inform based on those guidelines ensures that the research is always current.
3. Global application
The accurate assessment of biomarkers and the validity of resulting research findings depend on reliable DNA and RNA fusion panels and easily reproducible results. Data analysis and reporting in a comprehensive platform eliminates silos of valuable data and maximizes its application.
The SOPHiA DDM™ Platform enables the upload of multimodal data from any environment to one of the world’s largest networks of connected labs. Data remains the property of the healthcare institution, but pseudonymized and pooled with like data, it can propel research and ultimately treatment forward with the goal of improved patient care.
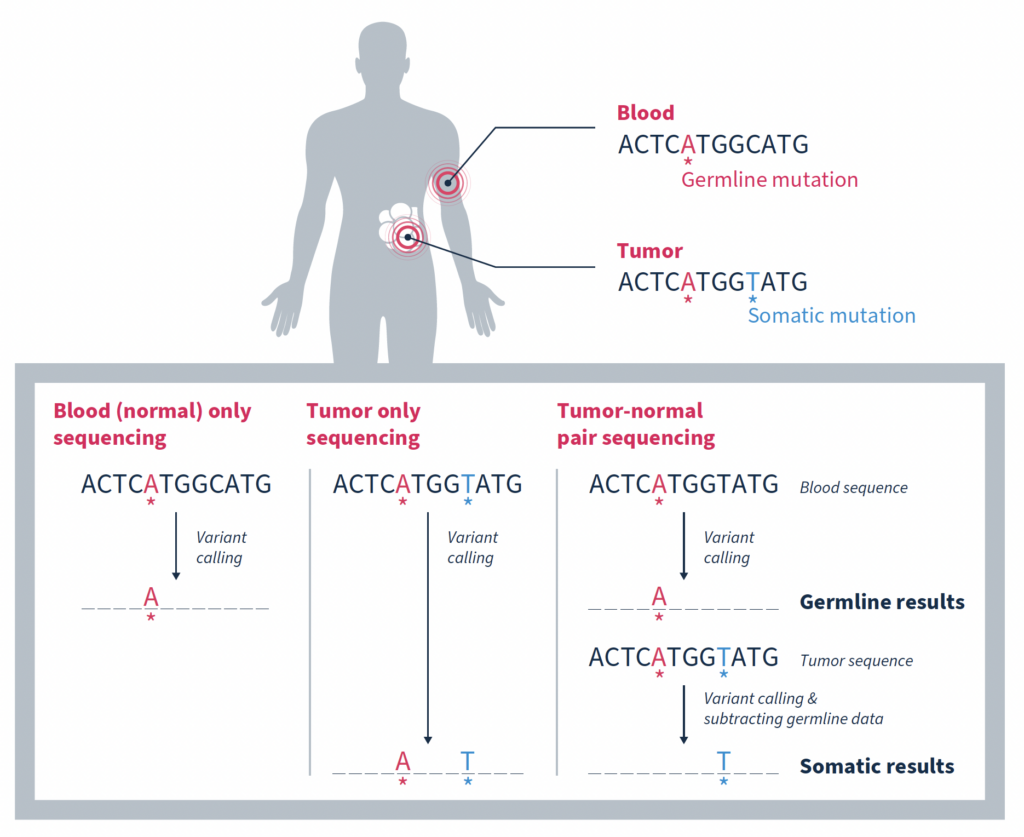
In the era of precision oncology, it has become increasingly common for patients diagnosed with cancer to undergo tumor sequencing. Identifying the mutations that make up a tumor’s genomic landscape can help guide selection of targeted therapies and inform prognosis. Despite the recognized value of tumor-only sequencing, labs performing this type of testing face a number of technical challenges that, if not properly addressed, can render the results uninformative or even misleading.
Although there are a variety of inherent challenges in tumor-only sequencing, all ultimately impact the ability to accurately distinguish somatic mutations driving tumorigenesis from germline variants associated with cancer predisposition. In fact, it has been estimated that as many as one third of mutations identified by tumor-only sequencing may be false-positive germline changes, including in potentially actionable genes1. Having an accurate picture of a tumor’s genomic makeup and contextual genetic environment is crucial to an accurate clinical assessment, which impacts therapeutic recommendations and represents the patient’s best chance for successful treatment.
In this blog we explore different strategies for enriching tumor analysis for somatic mutations and discuss why matched tumor-normal sequencing has become the preferred method.
Filtration by variants in large population databases
One approach is to use variants present in large population databases as a filter to remove likely germline variants from a tumor sample2. While this practice is generally effective, it will also remove true somatic variants that happen to be identical to germline variants, resulting in a false negative. Database-driven approaches can also overlook any rare germline variants missing from large population databases due to underrepresentation of non-White individuals. These variants will remain in the sequencing data and can result in false-positive germline findings.
Focusing on variants with low allele frequency
Taking allele frequency into consideration can help. This strategy is based on the premise that an allele frequency of 50% is consistent with a heterozygous germline variant, and an allele frequency of ~100% is consistent with a homozygous germline variant1. It then stands to reason that focusing on variants with a lower allele frequency increases the likelihood of somatic origin.
While this is true, such an approach can be complicated by many factors including contamination of the tumor sample with normal tissue, tumor heterogeneity, sequencing artifacts, difficulty mapping reads in regions of high homology, high level mosaic variants that arose early in differentiation, as well as changes in allele fraction due to copy number changes or loss of heterozygosity (LOH). Any of which can lead to inaccurate attribution of origin.
Matched tumor-normal sequencing that pairs analysis of a tumor sample with that of a comparable, normal sample – most often from the same individual – has been shown to be a more effective strategy, yielding more reliable identification of the somatic changes specific to a tumor1,3,4. As the name suggests, variants in the matched normal sample are determined to be germline in origin, or of alternate origin unrelated to the current tumorigenicity. When used as a filter against the tumor sample, somatic variants relevant to the cancer at hand can be identified with a high degree of confidence. Variants found at low frequencies in the normal sample can be confidently classified as false positives if they are not significantly enriched in the tumor.
Figure adapted from Mandelker, D, & Ceyhan-Birsoy, O. (2020)2.
While the most important function of matched tumor-normal sequencing is to identify and retain somatic mutations, it also serves other important functions.
Reducing false positives due to sample variability and sequencing artifacts
At the most simplistic level, biological samples can exhibit variability due to factors such as environmental influences, biological processes and sample handling. Matched-tumor normal sequencing provides a built-in baseline of background noise resulting from these factors, or from introduction of sequencing artifacts, that can be filtered out.
In the case of FFPE samples, extracted DNA is often fragmented and of a lower quality than fresh tissue samples. Matched tumor-normal sequencing provides a comparison that helps distinguish true alterations from noise resulting from degradation of the DNA, enhancing sensitivity.
Reducing false positives originating from CHIP variants
Cell-free DNA (cfDNA) samples, also known as liquid biopsy samples, contain DNA from tumor cells, but they also contain a significant amount of DNA from white blood cells. In many individuals, especially those who are older, these phenotypically normal blood cells contain acquired mutations subsequently increased in relative frequency due to clonal expansion. These clonal hematopoiesis of indeterminate potential (CHIP) variants often, but not always, occur in the same genes associated with blood cancers like leukemia. However, while they are indicative of an increased risk of developing a blood cancer in the future, they are not likely to be relevant to the tumor being analyzed.
Simultaneously sequencing matched white blood cells as a normal control can successfully distinguish somatic mutations that are relevant to driving tumorigenesis from somatic mutations arising from the normal process of clonal hematopoiesis4. This is such an important consideration that both ESMO and AMP guidelines specify that matched white blood cell sequencing should be used for interpretation of somatic variants in cfDNA testing5,6.
Removal of false positives arising from CHIP variants is not only important for accurate cfDNA analysis, but also FFPE analysis. In a study by Memorial Sloan Kettering Cancer Center (MSK) investigators, matched tumor-normal sequencing results showed that 5.2% (912/17,469) of patients with advanced cancer would have had at least 1 clonal hematopoietic (CH)-associated mutation erroneously called as tumor-derived in the absence of matched blood sequencing7. Of these CH variants, 49.7% of them were classified as oncogenic or likely oncogenic based on OncoKB™, and 3.2% were associated with approved or investigational therapies (e.g. mutations in IDH1/2). Failure to recognize such mutations as blood-derived may result in inaccurate precision therapy recommendations.
Streamlining germline variant analysis
The ability to distinguish between somatic and germline variants has the additional benefit of streamlining analysis of germline variants which have additional implication for a patient’s clinical care. Notably providing information about future disease risk which can be managed in part through surveillance as well as allowing for testing of family members who may also be at risk for disease.
It is for the reasons discussed here that MSK-ACCESS® powered with SOPHiA DDM™ for liquid biopsy and MSK-IMPACT®powered with SOPHiA DDM™ for comprehensive genomic profiling (CGP) utilize the matched tumor-normal analysis strategy to accurately delineate somatic variants from germline and CHIP variants.
Contact us to learn more about adopting advanced liquid biopsy and CGP technology in your laboratory.
References
Jones, S, et al. Sci Transl Med. 2015. 7(283):283ra53.
Mandelker, D, & Ceyhan-Birsoy, O. Trends Cancer. 2020;6(1):31-39.
Cheng, D.T, et al. J Mol Diagn. 2015;17(3):251–264.
Brannon, A.R, et al. Nat Commun. 2021;12:3770
Pascual, J, et al. Ann Oncol. 2022;33(8):750-768.
Lockwood, C.M, et al. J Mol Diagn. 2023;25(12):876-897.
Ptashkin, R.N, et al. JAMA Oncol. 2018;4(11):1589–1593.
Unleashing the power of healthcare data with the New Generation SOPHiA DDM™ Platform.
From Complexity to Clarity: The Critical Role of Data in addressing modern healthcare needs
As the burden of cancer and rare diseases continues to grow globally, the complexity of the diseases demands more sophisticated solutions. Researchers and clinicians are constantly striving to develop novel, more effective therapies, and diagnostic tools to improve patient outcomes and resolve the biggest unmet needs in global healthcare. At the core of these efforts, there is one key element: Data.
From diagnosis to therapy selection and drug development, data is now indispensable for diagnosis and personalized treatment. The rise of precision medicine highlights the critical need for cutting-edge solutions that can harness and analyze vast amounts of healthcare data, driving advanced decision-making to improve patient outcomes at scale.
In response to this pressing need, platforms like SOPHiA DDM™ have emerged as revolutionary solutions in advancing data-driven medicine. Since its initial release in 2015, SOPHiA DDM™ has pioneered how healthcare professionals use data, having analyzed over 1.8 million genomic profiles to date and accelerating the practice of precision medicine worldwide. With nearly 30,000 analyses per month, the SOPHiA DDM™ Platform has proven itself to be a vital tool in the fight against cancer, rare and inherited diseases. Yet, as healthcare evolves, so too must the tools and technologies that support it.
The Evolving Landscape of Healthcare Data
In recent years, the healthcare landscape has witnessed a dramatic increase in both the volume and complexity of data. Genomic, radiomic, and clinical data have become integral to understanding diseases on a deeper level. However, the ability to process, integrate, and analyze these diverse data sources remains a significant challenge for clinicians and researchers. This challenge highlights an unmet need in global healthcare: the necessity for platforms that can break silos within and among healthcare institutions, and bridge the gap between data generation and actionable insights, allowing for more accurate diagnoses and personalized treatment strategies.
Accelerating Innovation in Oncology and Rare Diseases
In response to this emerging need, SOPHiA GENETICS has just revealed the New Generation SOPHiA DDM™ Platform, aiming to stay at the forefront of precision medicine and address today the healthcare needs of tomorrow.
The New Generation SOPHiA DDM™ Platform not only enhances the speed and efficiency of data processing but also offers a powerful, web-based architecture designed to meet the evolving demands of clinical research. By leveraging advanced technologies like cloud computing and GPUs from world-class industry partners such as NVIDIA and Microsoft, SOPHiA DDM™ is set to revolutionize how healthcare professionals manage and interpret complex datasets to make informed decisions.
How does the SOPHiA DDM™ Platform empower 780+ global healthcare institutions worldwide to revolutionize their workflows?
Leveraging the groundbreaking capabilities of the SOPHiA DDM™ Platform, healthcare professionals benefit from significantly reduced turnaround times, enabling quicker insights from data upload to final analysis.
Moreover, the platform’s enhanced computing capabilities allow it to process larger and more complex datasets, paving the way for new applications such as Whole Genome Sequencing (WGS), Minimal Residual Disease (MRD), Liquid Biopsy, and more, providing deeper insights into the genetic underpinnings of diseases, helping clinicians tailor therapies to individual patients with greater precision.
In addition to genomics, the platform offers advanced multimodal analytics, which are essential for understanding diseases like cancer, where multiple data types (genomic, radiomic, and clinical) need to be integrated for a more comprehensive view of the patient’s condition and unique biology. This multimodal approach allows for the analysis and interpretation of diverse data across different modalities, leading to more accurate predictions and personalized treatment plans.
New Generation SOPHiA DDM™ Platform: One platform, multiple data modalities
One of the most significant advancements in the New Generation SOPHiA DDM™ Platform is its ability to offer genomic, radiomic, and multimodal analyses within a single, integrated workspace. This unified approach empowers healthcare providers to select the tools and applications that best suit their needs, whether they are focused on identifying genetic mutations, analyzing medical images, or integrating various data sources for predictive modeling.
Genomics Module: Ingests genomic data (FASTQ and VCF) to efficiently detect and annotate oncogenic or pathogenic variants, aiding in the precise classification of cancer mutations.
Radiomics Module: Processes imaging data (CT, MRI, PET-CT) to identify and segment areas of interest, extracting critical radiomic features for further analysis. This allows for the integration of imaging data with genomic insights, creating a more comprehensive view of a patient’s cancer.
Multimodal Module: Aggregates data from the genomics and radiomics modules alongside clinical, biological, and histological data. This holistic approach enables the training and validation of predictive models, providing a personalized, data-driven roadmap for patient care.
By integrating these diverse data types, the New Generation SOPHiA DDM™ empowers clinicians to make better-informed decisions, improving the precision of diagnosis and treatment in oncology, rare and inherited diseases.
Democratizing Data-Driven Medicine, Together
One of the key challenges in modern healthcare is the fragmentation of data. In many systems, vital information is siloed across different platforms and institutions, limiting the ability to generate a comprehensive understanding of a patient’s condition. SOPHiA GENETICS addresses this issue by promoting a decentralized, technology-agnostic, global platform where data can be securely shared among users, breaking down barriers to knowledge and experience exchange.
As Dr. Zhenyu Xu, Chief Scientific Officer at SOPHiA GENETICS, explains, “Our decentralized, multimodal analytics platform supports customers and helps break data silos by creating a global community where knowledge is safely and securely shared amongst users. The new generation of our SOPHiA DDM™ Platform is revolutionizing the user experience by blending our powerful AI algorithms with multimodal data to produce meaningful insights to further the field of precision medicine.”
As cancer therapies and data technologies continue to evolve, platforms like SOPHiA DDM™ will play a central role in shaping the future of precision medicine. The need for innovative, data-driven solutions is more urgent than ever, as healthcare providers strive to keep pace with the complexities of modern diseases.
Abhimanyu Verma, Chief Technology Officer at SOPHiA GENETICS, reflects on the broader impact of these advancements: “We pride ourselves on adapting our technology to meet our customers’ needs. As the technology infrastructure at most healthcare organizations worldwide has evolved, we are thrilled to continue to provide best-in-class technology and set them up for success. This new generation of our platform will allow us the flexibility to respond quickly to our customer’s evolving needs and introduce new features faster, and more efficiently.”
With its innovative architecture and advanced analytics capabilities, the new SOPHiA DDM™ Platform represents a major leap forward in precision medicine. By addressing the unmet needs in global healthcare data analysis, SOPHiA GENETICS is helping to pioneer a future where data-driven insights lead to more personalized, effective, and timely care for patients around the world.
Learn more about the New Generation SOPHiA DDM™ Platform here. Interested in getting a free demo of the Platform? Book it here!
The transition from the In Vitro Diagnostic Directive (IVDD) to the In Vitro Diagnostic Regulation (IVDR) in the European Union marks an important advancement in regulatory standards for genetic testing and analysis. The new standards promote transparency and traceability throughout genomic analysis processes, helping to ensure the reliability and accuracy of diagnostic results and ultimately patient safety.
Everyone wants to ensure that genomic analysis is safe for patients. However, healthcare institutions face real challenges in transitioning from IVDD to IVDR. Particularly with the use of complex software solutions for genomic data analysis, many of which are designated as research use only (RUO).
One of the biggest changes for healthcare institutions is that IVDR specifically regulates in-house manufactured tests. With few exceptions, healthcare institutions utilizing in-house manufactured tests must now meet the same requirements and proof of conformity with IVDR as manufacturers. These requirements extend to the software that is used for the analysis, interpretation and reporting of NGS data. Software developed in-house, including from public domain materials, must meet many of the same requirements and proof of conformity as commercial software solutions.
Here we answer some of the most pressing questions about what IVDR compliance entails for healthcare institutions performing genomic analysis and how CE-IVD certified software solutions can help.
Q: How does IVDR impact the analysis, interpretation, and reporting of NGS data?
A: Software used to support the analysis, interpretation and reporting of NGS data from genetic testing must conform to IVDR’s general safety and performance requirements (GSPR) to ensure reliability and safety.
Q: What are the key requirements for meeting IVDR compliance?
A: Genetic tests and their analytical software require that a healthcare institution perform the following to ensure IVDR compliance:
Implement a Quality Management System (QMS). The QMS must use standardized procedures to ensure that staff document, validate and monitor the effectiveness of the genetic test, and by extension its analytical software, at all times.
Maintain technical documentation, including safety and performance summaries. For genetic tests, IVDR imposes strict requirements for analytical and clinical validity. Extensive validation against known standards and clinical data sets, along with adherence to relevant clinical guidelines is expected. For analytical software, technical documentation should show that it has been developed in accordance with state-of-the-art practices.
It is also important to ensure and demonstrate that suppliers are complying with applicable regulatory requirements.
After launch, post-market surveillance is required to monitor the safety and clinical performance of the test. Genetic tests require regulatory management of updates as well as reporting of any serious incidents, with the corrective actions taken.
Q: When does IVDR take effect?
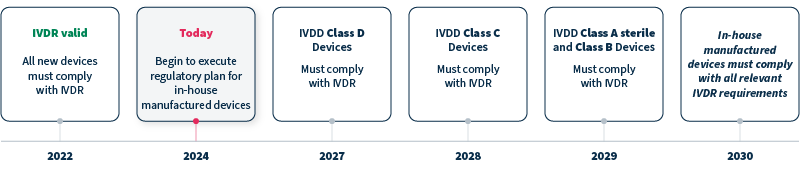
A: IVDR replaced IVDD in 2022, with timelines for compliance differing for devices with new, versus legacy, status and class. Genetic tests and analytical software that are Class C devices, for example, must comply with IVDR by 2028, and in-house manufactured devices must comply with all relevant IVDR requirements by 2030 at the latest (see timeline).
Q: How can commercial software solutions help with the IVDR transition?
A: When using a CE-IVD commercial software solution, the manufacturer’s CE-IVD certification and their existing technical documentation provide proof of compliance with current regulations, helping to lessen the burden for the healthcare institute.
Q: Can research use only (RUO) solutions be used to meet IVDR requirements?
A: RUO solutions are not intended or validated for clinical diagnostic use. RUO solutions will not be considered compliant without additional testing and validation as part of an in-house manufactured device.
Q: What are the most important considerations when evaluating commercial CE-IVD analytical software solutions?
A: When evaluating analytical software solutions, regulatory compliance is a must. However, it is also important to consider analytical and clinical validity. Does the solution provide reliable data processing and variant calling with high sensitivity and specificity? What variant types does it cover? Is it able to interpret variants according to established guidelines? Looking ahead to the future, can the solution scale with growing genomic analysis volumes?
As important as the new IVDR standards are to ensuring patient safety, it is clear that the transition poses a number of challenges to healthcare institutions performing genomic analysis. At SOPHiA GENETICS we’re proud to help simplify the transition, offering fast, reliable CE-IVD oncology applications powered by SOPHiA DDM™.
Dombrink, I et al. Critical Implications of IVDR for Innovation in Diagnostics: Input From the BioMed Alliance Diagnostics Task Force. Hemasphere. 2022. 6(6): e724.
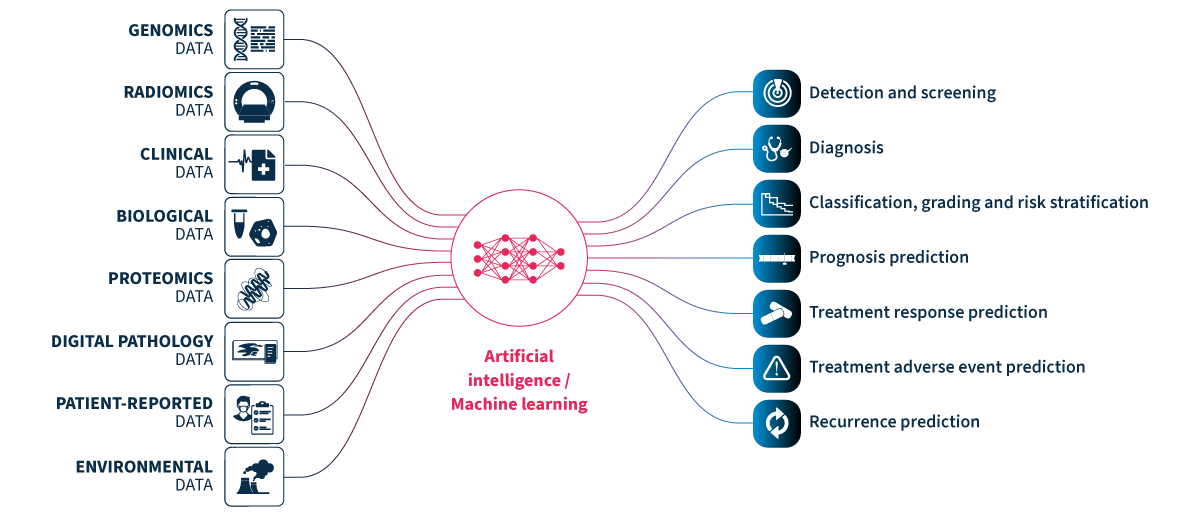
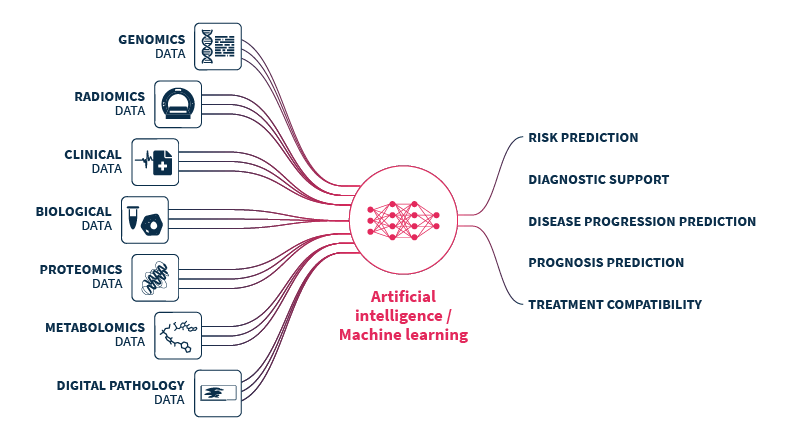
Multimodal healthcare datasets synergistically integrate diverse data modalities such as genomic, clinical, radiomic, proteomic, and biological data, to provide comprehensive insights into human biology and medical conditions. Multimodal datasets have the potential to predict outcomes more accurately and informatively than the sum of their parts (Fig. 1).
Figure 1.Multimodal healthcare data integrated and analyzed by artificial intelligence (AI)/machine learning can provide useful information for healthcare professionals to use to improve patient care. Genomics data. Radiomics data include x-rays, CT scans, MRI scans, ultrasound images, and mammograms. Clinical and biological data from electronic health records include patient histories, demographics, notes, diagnosis codes, procedure codes, laboratory results, and vital signs. Proteomics data. Digital pathology data. Patient-reported data includes questionnaires and health journals, as well as data from wearable devices monitoring heart rate, sleep patterns, and activity levels, and implantable devices such as pacemakers, insulin pumps, and continuous blood glucose monitors. Environmental data includes air quality and location data.
How are multimodal data and artificial intelligence (AI) advancing healthcare?
New data-driven technologies powered by novel ways of linking and analyzing patient data are set to transform the way that healthcare is delivered.1 Healthcare professionals routinely make use of multiple sources of data to arrive at a diagnosis and to decide on patient management.2 However, a significant level of expertise is required for an in-depth understanding of even a single data type (e.g. radiological images) such that it is unfeasible for individual healthcare professionals to master all areas. AI/machine learning technologies can be leveraged to bring together and analyze multimodal healthcare data, breaking data silos and creating robust and accurate predictive models.3 With the appropriate guidance around decision-making and communication, the valuable insights gained from these predictive models have the potential to support healthcare professionals to improve patient care.
Machine learning technologies can integrate data from disparate multimodal sources to provide a holistic understanding of patients’ health and medical conditions. Data are combined from multiple modalities with the aim of extracting complementary information to power predictive models that can find relationships between different variables/features that are not clearly visible or known by healthcare professionals. Indeed, multimodal data fusion models have consistently shown to provide increased accuracy (1.2-27.7% higher) and performance (AUC 0.02-0.16 higher) than models that utilize data from single modalities for the same task.4
Oncology is one of the medical specialties that most commonly leverages multimodal methods for clinical decision support.5 Machine learning technologies have the potential to explore complex and diverse data to support healthcare professionals from screening to treatment (including relapse).6 Identification of risk factors can support non-invasive patient screening and preventive care.3 Detection of patterns in easily accessible data can help identify diagnostic or prognostic biomarkers to improve patient risk stratification or selection for clinical trials. Identification of predictive signatures of risk factors, adverse treatment reactions, treatment responses, or treatment benefit, can guide decisions around patient management.
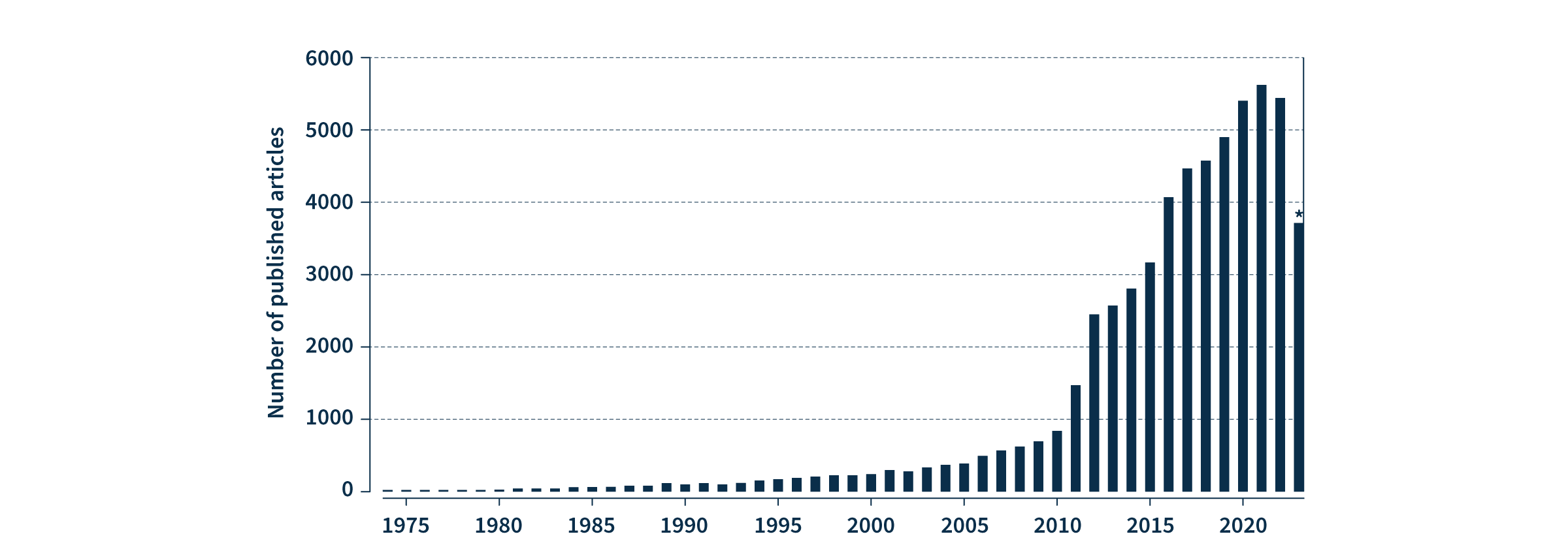
Figure 2. The number of PubMed articles published on multimodal oncology data has dramatically increased in recent years. PubMed search for ((multimodal) AND (oncology)) OR ((multimodal) AND (cancer)). *2023 analysis includes data available at time of writing (January-September).
With data privacy and security paramount, multimodal healthcare data can also be leveraged to accelerate advances in medical research, such as the discovery of novel biomarkers and therapeutic targets for drug development, as well as supporting population health management by providing a comprehensive view of health trends and outcomes. The rapid increase in peer-reviewed publications on the topic over the last 13 years demonstrates that the extraordinary value of multimodal oncology data is already recognized by the scientific and medical communities (Fig. 2). Leveraging machine learning to collate and analyze the vast diversity of multimodal data for data-driven precision medicine is on track to drive the next revolution in healthcare.
Data-driven insights with SOPHiA DDM™️ multimodal healthcare analytics
SOPHiA DDM™ multimodal healthcare analytics will have the potential to break data silos by streamlining the integration of longitudinal oncology data from multiple sources and modalities – including but not limited to genomic, radiomic, digital pathology, biological, and clinical data. The SOPHiA DDM™ Platform uses machine learning-powered analytics to assemble, standardize, and transform multimodal data into accessible data-driven insights, facilitating the identification of multimodal predictive signatures, as well as treatment response patterns and trends. To learn more and get in touch, visit the webpage.
Product in development – Technology and concepts in development. May not be available for sale.
Glossary
Area under the ROC curve (AUC) – A ROC (receiver operating characteristic) curve is a graph that plots true and false positive rates to demonstrate the performance of a model. AUC measures the area underneath the ROC curve to provide an aggregate measure of performance. AUC values range between 0 and 1, with a score of 0 meaning that all predictions are wrong, and a score of 1 meaning that all predictions are 100% correct. Essentially, AUC represents the probability that a positive result is truly positive and a negative result is truly negative.
Omics data – Large-scale information related to the biology of organisms.
Digital pathology images – Scanned images of tissue samples on glass slides.
Lipkova J, et al. Cancer Cell. 2022 Oct 10;40(10):1095-1110.
Huang SC, et al. NPJ Digit Med. 2020 Oct 16;3:136.
Kline A, et al. NPJ Digit Med. 2022 Nov 7;5(1):171.
He X, et al. Semin Cancer Biol. 2023 Jan;88:187-200.
What is liquid biopsy?
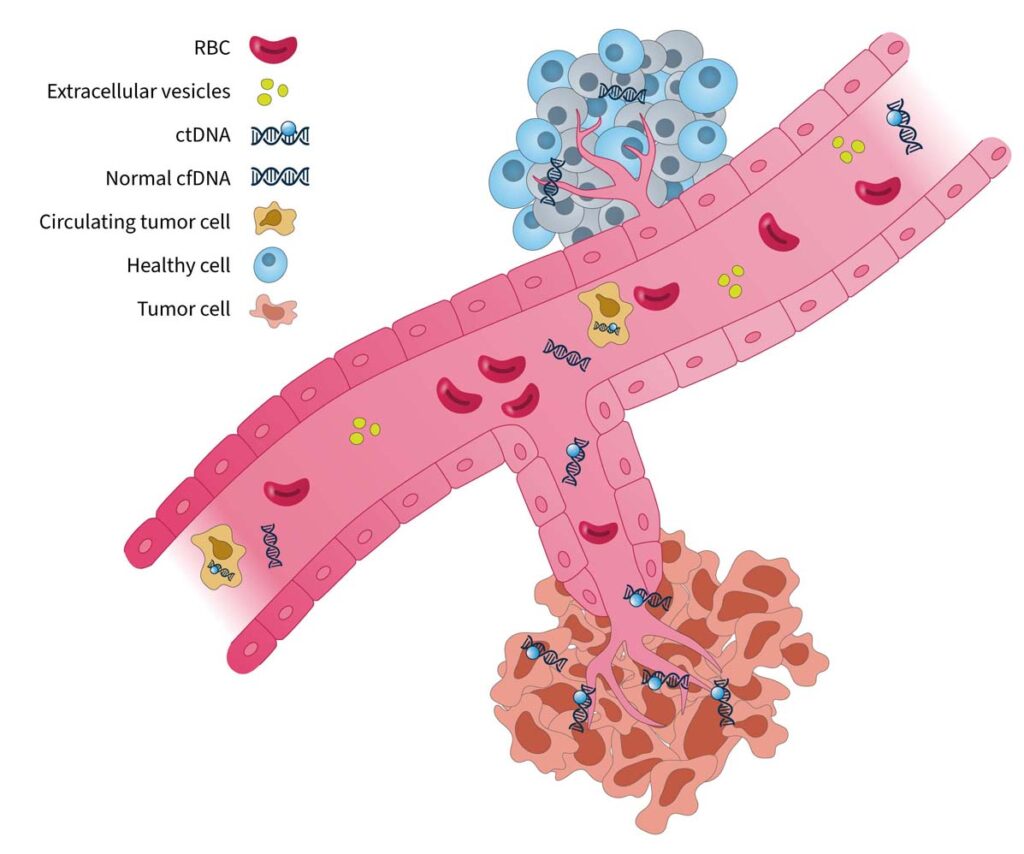
Liquid biopsies enable analysis of biofluids, typically blood, to examine biomarkers shed by solid tumors into circulation1. They can detect actionable genomic alterations in a non-invasive way, providing valuable insights to facilitate early cancer detection and disease monitoring2.
Tumor-derived biomarkers that are a source for liquid biopsy analysis include (Fig. 1):
Circulating tumor cells (CTCs)
CTCs are initially released from primary tumors in the tissue, travel through the bloodstream, and account for the development of metastatic tumors at distant sites in the body. As they are live cells, they have the potential to be used for functional analysis such as therapy sensitivity/resistance evaluation3. However, CTCs are rare events in the blood, which makes them difficult to identify and characterize in routine clinical practice1.
Extracellular vesicles (EV, i.e. microvesicles and exosomes)
EVs are membrane-enclosed structures containing proteins, genetic material, and lipids that can provide biological information on the cell of origin4. Due to their role in pathological processes, EVs are an attractive analyte for liquid biopsy, but their isolation and purification is technically challenging1.
Cell-free DNA (cfDNA) and circulating tumor DNA (ctDNA)
cfDNA refers to DNA fragments that are freely circulating in the bloodstream, primarily originating from normal cells5. Circulating tumor DNA (ctDNA) is the small portion of cfDNAthat derives from tumor cells or CTCs undergoing cell death (i.e. apoptosis or necrosis)5. There are well-established methods for isolating cfDNA, and for analyzing it using methods such as PCR and next-generation sequencing (NGS)-based tests6, making it an ideal and feasible substrate for routine genomic analysis.
Figure 1. Blood-based cancer biomarkers in liquid biopsy7.RBC, red blood cell.
The analysis of cell-free DNA is a promising method for guiding clinical decisions and can complement current standard-of-care practices8.
What are the clinical applications of liquid biopsy?
In the era of precision medicine, tumor molecular profiling is a critical tool to identify targetable alterations and guide treatment decision-making9. Tissue biopsy is currently the gold standard for tumor profiling8; however, there are limitations associated with this approach:
Surgery or biopsy to obtain tissue samples can be invasive, expensive, and time-consuming, making it unsuitable for repeat sampling and longitudinal disease monitoring10.
In certain cancer types, it can be difficult to obtain sufficient tissue quantity and quality10. For example, clinical studies show that up to 20% of non-small cell lung cancers (NSCLC) biopsies are inadequate for molecular analysis11,12.
The information provided by tissue biopsy is limited to a single point in space and time, failing to represent complex inter- and intra-tumor heterogeneity13.
Liquid biopsy has the potential to be a transformative tool in clinical oncology, showing promise for applications in many stages of cancer management (Fig. 2):
Liquid biopsy is non-invasive, easily repeatable, and relatively fast to perform8. This makes it an ideal approach for when tissue biopsy is not feasible, or a time-sensitive decision needs to be made.
It allows real-time monitoring of changes during the course of disease, providing insights into intra-patient spatial and temporal tumor heterogeneity8,13.
A major advantage of liquid biopsy is its ability to detect minimal residual disease (MRD)14, which is not detectable by conventional routine tests. MRD refers to the small number of cancer cells that remain in the body following treatment that can drive the reemergence of cancer14. Measurement of MRD using liquid biopsy can provide valuable insights into treatment response and risk of cancer recurrence14.
Figure 2. The advantages and clinical utility of liquid biopsy in the cancer care journey10–15.
Innovations in liquid biopsy analysis over the past decade have led to the regulatory approvals of blood-based tests to guide treatment for NSCLC, prostate, breast, and ovarian cancers16. Clinical guidelines have also provided expert recommendations for its use in specific clinical scenarios8,15. Despite great advances in technology and its increasing utility in clinical practice, there are still challenges to overcome when using liquid biopsy to identify clinically relevant information.
Overcoming “fisherman’s luck” in liquid biopsy
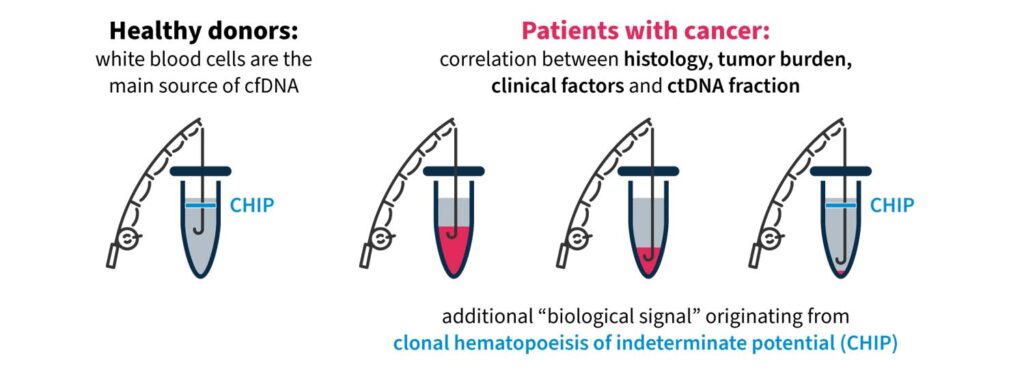
One challenging aspect of liquid biopsy analysis is that ctDNA concentration varies greatly across cancer types and between patients17. In patients with cancer, the quantity of ctDNA in the blood can be impacted by several factors, including histology, tumor site, clinical factors (age, sex, treatment history, etc.), and ctDNA fragmentation17. Therefore, it is important to have a robust test to detect clinically relevant variants, even at low ctDNA concentrations against a cfDNA background.
Another factor that may impact liquid biopsy analysis is the presence of clonal hematopoiesis of indeterminate potential (CHIP). In healthy individuals, the majority of cfDNA arises from hematopoietic cells (i.e. stem cells in the bone marrow that give rise to other blood cells)18. Normal hematopoietic cells accumulate somatic mutations during aging, known as CHIP, which are technically indistinguishable to tumor-specific mutations in NGS assays18,19. It is important that the biological noise caused by CHIP signals are removed in liquid biopsy analysis to eliminate false positive variant calls and give an accurate representation of disease burden19,20.
These biological confounders can make “fishing” for clinically relevant information in cfDNA a challenge (Fig. 3). For example, if a patient has high disease burden, there is likely more ctDNA available to analyze, which makes it easier to “catch” what you are looking for. However, if there is less ctDNA and more biological noise, you may need to modify your tools and approach to improve your yield.
Figure 3. “Fishing” for clinically relevant information in liquid biopsy can be complicated by biological confounders17,18.
Highly precise and sensitive liquid biopsy technologies are needed to overcome “fisherman’s luck” and detect rare, causative variants and disease burden in cfDNA. Guidelines issued by the ESMO Precision Medicine Working Group on the use of cfDNA assays in clinical practice discuss the need for advanced techniques capable of capturing spatial and temporal tumor heterogeneity and reducing rates of false negatives8.
Pioneer innovation with SOPHiA DDM™ for Liquid Biopsy
SOPHiA GENETICS is at the forefront of innovation in liquid biopsy technology for tumor profiling. The advanced proprietary algorithms of the SOPHiA DDMTM Platform empower clinical researchers to reveal deep genomic insights from cell-free DNA samples.
With a streamlined, sample-to-report NGS workflow, you can:
Retain custody of your cell-free DNA samples and your data: Analyze your samples directly in your lab, combining an in-house wet lab workflow with the cloud-based analytics of the SOPHiA DDM™ Platform.
Customize your application to focus on the biomarkers that matter most to you: Develop a high-quality application that suits your needs and receive comprehensive support at every step with the SOPHiA DDM™ MaxCare Program.
Reduce noise with our proprietary molecular barcoding technology: Detect variants at low frequency (0.5% VAF) starting from 25 ng cell-free DNA with our proprietary unique molecular identifier (UMI) technology, CUMIN™.
In addition, we are excited to be collaborating with Memorial Sloan Kettering Cancer Center (MSK) to decentralize MSK-ACCESS® for liquid biopsy, designed to provide a maximum coverage of cancer disease variants in ctDNA20. By combining MSK’s clinical expertise in cancer genomics, the predictive algorithms of the SOPHiA DDM™ Platform, and the power of the global SOPHiA GENETICS community, the collaboration aims to expand access to precision cancer analysis capabilities worldwide.
Read more about how you can enhance your analytical capabilities and advance your clinical research here.
Genomic instability is a hallmark of cancer and targeting its mechanisms has helped inform effective therapeutic strategies1,2. However, there are limitations with current methods of genomic instability assessment. Here, we explore genomic instability in the context of homologous recombination deficiency and the value of deep learning-based methods of detection.
What is genomic instability and how is it caused?
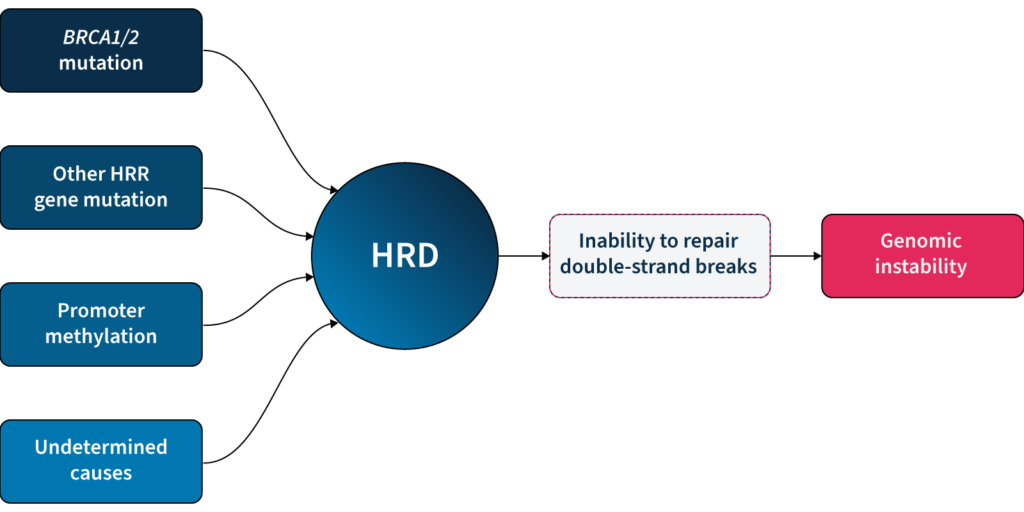
The DNA in our cells endure up to one million damaging events each day, caused by both exogenous (i.e. environmental) and endogenous (i.e. internal metabolic) factors3. These events activate a complex network of DNA damage response (DDR)pathways, which facilitate DNA repair and maintain the stability of the genome4. Mutations and/or dysfunction in DDR pathways can lead to unrepaired DNA damage, resulting in genomic instability4. Genomic instability, in turn, increases the cell’s propensity for genetic alterations that cause cancer initiation and progression4,5.
One of the major DDR pathways is the homologous recombination repair(HRR) pathway, responsible for repairing double-strand breaks (DSBs) in DNA5. Loss of function in HRR, known as homologous recombination deficiency (HRD), causes cells to rely on error-prone DNA repair pathways, resulting in the accumulation of genetic aberrations that lead to genomic instability5 (Fig 1). HRD is a well-established prognostic and predictive biomarker in different cancer types (e.g. ovarian, breast, prostate, and pancreatic)5–7.
Fig 1: Genomic instability can be caused by the inability of HRD cells to repair double strand breaks.
Why is it clinically relevant to measure genomic instability?
HRD-positive tumors are sensitive to targeted inhibition of poly-ADP ribose polymerase (PARP), key proteins involved in DSB repair7. By blocking PARP, the HRD-positive cell can no longer rely on error-prone pathways for DSB repair and the cell dies, a process known as ‘synthetic lethality’5,6. PARPi therapy has revolutionized the management of HRD-positive patients with advanced ovarian cancer, significantly improving progression-free survival when used as a first-line maintenance therapy8. PARPi therapies also have approved indications in breast, pancreatic and prostate cancer9, with trials underway in other cancer types, such as colorectal10.
Based on the predictive value of HRD status for PARPi benefit, clinical guidelines recommend HRD testing in patients with advanced ovarian cancer7,11,15. HRD status can be determined by examining 1) the underlying causes of HRD, and 2) the effect of HRD, i.e. genomic instability5,7. The most well-known causes of HRD are loss-of-function mutations in HRR genes, including BRCA1 and BRCA25,7. However, loss-of-function in HRR genes is diverse amongst patients12, making patient stratification solely based on genotyping challenging. Also, approximately 30–40% of HRD cases are due to unknown causes13,14. Measuring genomic instability allows the assessment of HRD, regardless of its underlying etiology5,7.
Genomic instability status can help identify a sub-group of women who are BRCA wild-type but may still derive benefit from PARPi therapy15. By measuring genomic instability, clinicians and researchers can therefore go beyond HRR mutation detection and expand the potential benefit of PARPi in patients.
How is genomic instability measured?
Many methods for measuring genomic instability rely on the identification of specific mutational signatures or genomic ‘scars’ associated with large-scale structural rearrangements in chromosomes. In HRD-positive cancers, the characteristic genomic scars are loss of heterozygosity (LOH), large-scale state transitions (LST), and telomeric-allelic imbalance (TAI)16–18.
Click the boxes below to learn more:
LOH
A cross-chromosomal event that results in loss of part of a gene or entire gene(s) and the surrounding chromosomal region.
LST
Chromosomal breaks between adjacent regions of at least 10 Mb.
TAI
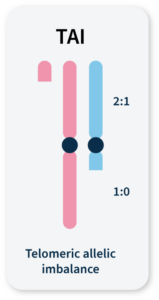
Accumulation of a discrepancy in the 1:1 allele ratio at the end of the chromosome (telomere).
The combined number of LOH, LST, and TAI events generate a genomic instability score (GIS) that reflects the level of genomic instability. Some commercially available HRD tests combine tumor BRCA mutation testing with a GIS5,19. Methods that integrate multiple genome-wide signatures (e.g. HRDetect) are among the most promising for detecting HRD status7,20. However, both GIS and HRDetect methods require deep genomic profiling data (>30x coverage), which can be costly and difficult to implement in routine analysis.
Alternative approaches that rely on the detection of copy number changes from WGS at low (~1x) sequencing depth (low-pass WGS) can predict tumor HRD status21,22 and provide an affordable and easy-to-implement HRD detection method. However, the sensitivity of existing methods that solely rely on this type of genomic scar to identify HRD samples is limited, and their utility in a clinical context remains untested22.
Unlocking the full potential of low-pass WGS in HRD detection requires going beyond the enumeration of biomarker events and examining alternative features of the cell that can result from genomic instability.
GIInger™: A deep learning approach to genomic instability measurement
To overcome the limitations of current genomic instability measures, our expert team at SOPHiA GENETICS developed the GIInger™ algorithm exclusively available on the SOPHiA DDM™ Platform. GIInger™ is a deep learning-based approach to measuring genomic instability in ovarian cancer samples. Rather than relying on the enumeration of biomarkers events, GIInger™ leverages differences in the spatial distribution of genomic scars in low-pass WGS coverage profiles23.
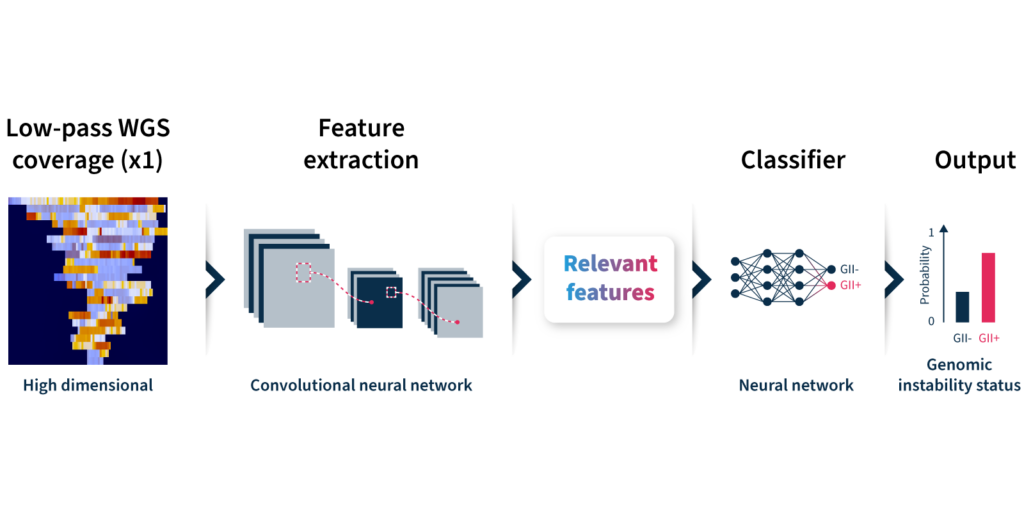
Let’s take a closer look at how the algorithm predicts genomic instability status (Fig 2):
Input: GIInger™uses coverage profiles from low-pass WGS data, requiring only ∼1x coverage depth (minimum ∼0.4x).
Convolutional neural network (CNN): Coverage profile features are extracted and passed through a CNN that has been trainedon a sample dataset to classify genomic instability.
Output: The GIInger™ model produces a Genomic Integrity Index (GII) which is predictive of a sample’s genomic instability status.
Unfamiliar with deep learning terminology? Read our guide on machine learning jargon.
Fig 2. Schematic of GIInger™ architecture.
By adopting GIInger™ into next generation sequencing (NGS) workflows, clinical researchers can benefit from an in-house, affordable approach to genomic instability measurement. The SOPHiA DDM™ Platform offers applications that enable laboratories to easily implement GIInger™ into their routine NGS analysis:
SOPHiA DDM™ HRD Solution (also available as CE-IVD-marked application): A HRD detection solution that combines targeted sequencing panel of 28 HRR genes, including BRCA1 and BRCA2, with GIInger™ in a single end-to-end workflow.
SOPHiA DDM™ GIInger Genomic Integrity Solution: A bioinformatic pipeline for prediction of genomic instability status in samples. Designed to analyze WGS data, this solution offers a flexible approach to genomic instability measurement that can complement capture-based BRCA assays for a complete HRD assessment.
Want to see how GIInger™ can help maximize insights from your data? Get in touch with our team and request a demo.
An endemic problem in the healthcare industry is that there are too few staff 1 to give all patients the time that they need to receive the best care
Precision medicine is set to revolutionize healthcare,2 and state-of-the-art technologies are essential to achieve this. These technologies will be used to detect patterns in large quantities of genetic, biological, clinical (research), and environmental data, to extract insights related to personalized patient care. Furthermore, an endemic problem in the healthcare industry is that there are too few staff1 to give all patients the time that they need to receive the best care. State-of-the-art technologies have the potential to streamline processes so that healthcare professionals (HCPs) have more time to dedicate to their patients.
When discussing state-of-the-art technologies, we inevitably come across terms like artificial intelligence (AI), machine learning (ML), deep learning (DL), and neural networks (NN). And although we might have a vague understanding of what each of these terms means, it is difficult to distinguish between them and to know how to speak about them when discussing their potential impact on healthcare with colleagues and collaborators. This guide will clarify these terms.
Artificial intelligence, machine learning, deep learning, or neural networks?
In a nutshell, artificial intelligence (AI) is when computer systems simulate human intelligence by performing tasks that typically require human cognitive abilities.
Machine learning (ML) is one of the key techniques used in AI, where algorithms and statistical models are designed by humans to enable computer systems to learn and improve from experience without being explicitly programmed.
Deep learning (DL) is a subset of ML where algorithms use artificial neural networks to process and analyze large amounts of data, extracting relevant features and patterns.
Neural networks (NN) are a fundamental component of DL algorithms, which are designed to simulate the behavior of the human brain with a network of interconnected nodes (or neurons) that process and transmit information.
For more detailed explanations, click the terms below to reveal.
Artificial Intelligence
AI is a field of computer science that aims to create intelligent machines that can perform tasks such as visual perception, speech recognition, decision-making, and natural language understanding. AI systems use algorithms and statistical models to process large amounts of data, identify patterns, and make predictions or recommendations based on the data.
Although the term AI was coined in 1956 by John McCarthy, the possibility that machines could simulate human behavior and “think” was raised earlier by Alan Turing in 1950.3 Since then, computational power has grown exponentially, and AI is integrated into our daily lives in many forms. For example, many of us use (the likes of) Siri, Alexa, or Google Assistant without a second thought.
More recently, AI has become part of medical practice, where it can improve patient care by speeding up processes and achieving greater accuracy.3 Radiological images, pathology slides, genetic information, and patients’ electronic medical records can be evaluated using AI to aid with administrative tasks and diagnosis and treatment decisions, enhancing the capabilities of clinicians.
Machine Learning
ML is an AI technique for fitting models to data that involves the development of algorithms and statistical models.4 The machine “learns” by training models with data to identify patterns so that it can make predictions or decisions. Machine learning is a widely used AI technique and forms the basis of many approaches within the field.
In healthcare, the most common application of ML is in precision medicine, where patient data are used to predict which treatment strategies are most likely to succeed.4 In order to make predictions, the algorithms generally require a training dataset for which the outcome variable (e.g., onset of disease) is known – this is called supervised learning.
Deep Learning
The most complex forms of machine learning involve DL, or NN models, which have many layers of features or variables that predict outcomes.4 The more layers a network has, the deeper it is, hence the term “deep learning”. The improved capabilities of today’s graphics processing units and cloud architectures make it possible to process and analyze thousands of hidden layers of features.
In healthcare, pattern recognition through DL involves teaching a computer what certain groups of symptoms or radiological images, for example, look like via repetitive algorithms.5 The algorithms enable the computer to learn and improve from experience by adjusting weights and biases. An example of this is when Google’s artificial brain project trained itself to recognize cats based on 10 million YouTube videos, with recognition efficiency improving the more images it reviewed.
DL algorithms can be used for a wide range of applications, such as image and speech recognition, natural language processing, and autonomous systems. A common application of DL in healthcare is for the identification of clinically relevant features (e.g., tumors) in imaging data, which may not be perceived by the human eye.4 Using DL to analyze radiology images can provide a more accurate diagnosis than the previous generation of automated tools for image analysis, computer-aided detection (CAD).
Neural networks
NNs are a fundamental building block of DL algorithms, which can learn and make decisions by themselves.6 They are an interconnected network of nodes (neurons) that mimic the human brain, with weighted communication channels between them.7 Each neuron receives input from other neurons in the previous layer, applies a mathematical operation to that input, and then passes the output to neurons in the next layer. One neuron can react to multiple stimuli from neighboring neurons and use weights and biases to adjust the strength of connections between them. The whole network can change its state based on different inputs received from the environment. As a result, NNs can generate outputs in response to environmental input stimuli, just like the human brain reacts to the environment around us.
How are artificial intelligence and machine learning used in healthcare?
AI and ML can be used in healthcare to assist HCPs in streamlining processes, reducing costs, and perhaps, most importantly, making faster, data-driven clinical decisions, all with the aim of improving patient outcomes. Advances in big data analytics using AI techniques are unlocking clinically relevant information hidden in increasingly available healthcare data, which is successfully assisting HCPs with clinical decisions.8 There are five key areas in which AI and ML are currently accelerating healthcare – predictive medicine and imaging, patient support, services management, physical assistance, and drug development.
Predictive medicine and imaging – supporting quicker and better-informed decision-making
ML has the potential to analyze individual patient data to predict risk, support diagnosis, predict disease progression and prognosis, and to identify the most appropriate treatment regimens.9 In addition, ML has the potential to identify risk factors and drivers for each patient, to help target healthcare interventions for better outcomes.
ML has primarily been used in healthcare to analyze data from imaging, genetic testing, and electrodiagnosis.8 These data are analyzed by AI technologies to cluster patient traits and associate them with a diagnosis, or to predict disease outcomes or response to treatment.
The application of ML to medical imaging has been found to improve accuracy, consistency, and efficiency. In 2017, Arterys developed the first US FDA-approved clinical cloud-based DL application, CardioAI.6 CardioAI analyzes cardiac magnetic resonance images (MRIs) to provide information such as cardiac ejection fraction in a matter of seconds, and has since expanded to cover additional organs and imaging techniques. The time-saving implications from introducing AI support platforms into clinical practice can be quite substantial; radiologists can save ~1 hour per day interpreting chest CTs,10 DL can measure pediatric leg lengths 96x faster than subspecialty-trained pediatric radiologists,11 and AI systems can automate the triaging of adult chest radiographs.12
ML has successfully been used to screen for diabetic retinopathy, identify nonmelanoma and melanoma skin cancers, predict seizures, predict bladder volume, predict cardiovascular risk, and predict progression of Alzheimer’s disease and response to drug therapy.6,7
Patient support – increasing independence and streamlining outpatient care
There are multiple ways in which AI has been used to provide outpatient care. In combination with robotics, AI has been harnessed to restore movement control in patients with quadriplegia for example, and to control prostheses.8 Rehabilitation robots can physically support and guide a patient's limb(s) during physical therapy.9 AI can also be used to assist the independent living of elderly and disabled people with tools such as fall detection systems and wheelchairs controlled by facial expressions.7
AI-powered virtual assistance like chatbots and voice assistants can provide patients with personalized medical advice and support from home, helping patients to manage their own health more effectively with the aim of reducing the workload of HCPs.4,5 Furthermore, wearable systems can support continuous patient monitoring and healthcare delivery.9
Health services management – freeing up more time to spend with patients
AI can support HCPs to work more efficiently, freeing up more time to spend on patient care. AI systems can provide HCPs with real-time medical information updates, coordinate information tools for patients, optimize logistics processes, benchmark data for analyzing services delivered, and much more.9 Process automation, specifically, can be leveraged for tasks such as claims processing, clinical documentation, revenue cycle management, and medical record management.4 Ultimately, the administrative assistance provided by AI creates more time for human interactions.
Physical assistance – increasing precision and efficiency
AI has the potential to transform surgical robotics through devices that can perform semi-automated surgical tasks with increasing efficiency.9 AI technologies can guide surgical tools and make more precise movements than possible within our capacity as humans.5
Drug development – accelerating development and cutting costs
AI techniques streamline the design and development of new drugs by analyzing the vast amount of data available from clinical trials and databases to identify new drug targets and predict drug efficacy and safety.9
How will AI and machine learning impact the future of healthcare?
AI and ML are set to drive the future of healthcare. In particular, ML is a key component in the advancement of precision medicine.4 AI and ML will greatly enhance risk prediction and diagnosis of diseases, and will facilitate personalized treatment strategies based on a broad spectrum of individual patient characteristics. It seems feasible that most radiology and pathology images will soon be examined by a machine and that AI and ML will help HCPs to remotely monitor patients. Speech and text recognition are already used for patient communication and to capture clinical notes, and their usage is likely to increase. AI and ML will also help to accelerate and reduce costs associated with the drug development process.
The successful integration of AI and ML technologies into healthcare requires more than just reliability and accuracy. Several critical factors must be in place to ensure sustainable adoption, including integration with electronic health record systems, standardization, adequate funding, improved regulatory approval processes, staff training, and continuous algorithm optimization with new data.
However, the most crucial factor is transparency. The complexity of AI/ML algorithms and models can make them difficult to interpret or explain, potentially raising concerns about accountability, trust, and privacy. To promote the responsible and sustainable adoption of these technologies, healthcare institutions and regulatory bodies must first establish governance mechanisms and monitoring structures to safeguard the interests of providers and patients alike. This will not only ensure a smooth transition but also foster trust in the use of AI technologies in healthcare.
It seems increasingly clear that AI systems will not replace human HCPs but will instead enhance their capabilities to improve the care of patients. It is important that HCPs are trained and provided with the skills to efficiently work alongside AI. The goal will be to balance a mutually beneficial relationship that leverages on the speed and analytic potential of AI with the uniquely human strengths of empathy, nuance, and seeing the big picture.
Conclusion
Whether AI, ML, DL, or NN, the overall field of artificial intelligence is booming, and the possibilities for improving healthcare are exciting. Although it may seem fantastical, HCPs are already working alongside machines to enhance clinical decision-making, and the future in this field has huge potential for improving patient care.
At SOPHiA GENETICS, we have a strong background in developing ML algorithms that aim to extract actionable insights from genomic data. SOPHiA DDM™ for Multimodal (product in development) leverages on propriety ML algorithms, with the potential to maximize the value of multimodal health data. Our technology has so far been utilized in retrospective research studies to assess prognosis and predictive biomarkers in non-small cell lung cancer (NSCLC),13 investigate new meningioma biomarkers to further understand treatment response patterns,14 evaluate pathological complete response status and treatment response in patients with early triple negative breast cancer (TNBC),15 and estimate the risk of disease upstaging, disease-free and overall survival in kidney cancer.16 These studies report data associated with products or concepts in development. They are not available for sale and not intended for use in diagnostic procedures or treatment decisions.
To learn more about SOPHiA DDM™️ Multimodal Healthcare Analytics for the visualization of longitudinal patient data and cohorting, explore here or request a demo here.
References
Boniol M, et al. BMJ Glob Health. 2022;7(6):e009316.
Johnson KB, et al. Clin Transl Sci. 2021;14(1):86-93.
Mintz Y, Brodie R. Minim Invasive Ther Allied Technol MITAT Off J Soc Minim Invasive Ther. 2019;28(2):73-81.
Davenport T, Kalakota R. Future Healthc J. 2019;6(2):94-98.
Amisha et al. J Fam Med Prim Care. 2019;8(7):2328-2331.
Kaul V, et al. Gastrointest Endosc. 2020;92(4):807-812.
Rong G, et al. Engineering. 2020;6(3):291-301.
Jiang F, et al. Stroke Vasc Neurol. 2017;2(4).
Secinaro S, et al. BMC Med Inform Decis Mak. 2021;21(1):125.
Yacoub B, et al. Am J Roentgenol. 2022;219(5):743-751.
Zheng Q, et al. Radiology. 2020;296(1):152-158.
Annarumma M, et al. Radiology. 2019;291(1):196-202.
Ferrer L, et al. J Clin Oncol. 2022;40(16_suppl):8542-8542.
Graillon T, et al. Neuro-Oncol. 2021;23(7):1139-1147.
Groheux D, et al. J Clin Oncol. 2022;40(16_suppl):601-601.
Boulenger de Hautecloque A, et al. J Clin Oncol. 2022;40(16_suppl):4547-4547.
The HGVS nomenclature guidelines are used worldwide for genetic variant interpretation but can seem complicated and difficult to understand and apply. That is why we have created this beginner’s guide to mutation nomenclature using the HGVS recommendations, with clear visual examples that break down the process into bitesize pieces.
The Human Genome Variation Society (HGVS) nomenclature standard was developed to prevent the misinterpretation of variants in DNA, RNA, and protein sequences. The HGVS nomenclature standard is used worldwide, especially in clinical diagnostics, and is authorized by the Human Genome Organisation (HUGO).1,2
HGVS General Terminology Recommendations1
X Do not use
✔️ Recommended terminology
Mutation or polymorphism
Variant, change, allelic variant Can be used for cancer tissue: Mutation load and tumor mutation burden
HGVS follow recognized standards for the nomenclature of DNA and RNA nucleotides, the genetic code, amino acid descriptions, and cytogenetic band position in chromosomes.3
2. How to read mutation nomenclature: Breaking down the variant description
The HGVS recommendations for mutation nomenclature state that the format of a complete variant description should first include the reference sequence, followed by the variant description, and then the predicted consequence in parentheses. For example, NM-004006.2:c.4375C>T p.(Arg1459*) (Figure 1).
Figure 1. Application of the HGVS nomenclature recommendations for sequence variants
2.1. How to read mutation nomenclature: Reference Sequence
The HGVS nomenclature recommendations for sequence variants state that a complete variant description should begin with the reference sequence.1 The reference sequence accession number begins with a two-letter abbreviation (explained in Table 1), followed by a multi-digit number, and finally a version number.
Table 1. Meaning of the two-letter abbreviation at the beginning of a reference sequence accession number.
Abbreviation
Reference sequence based on a:
NC
Chromosome
NG
Gene or genomic region
LRG
Locus Reference Genomic sequence: Gene or genomic region, used in a diagnostic setting
NM
Protein-coding RNA (mRNA)
NR
Non-protein-coding RNA
NP
Protein (amino acid) sequence
2.2. How to read mutation nomenclature: Description of variant
The variant description begins by depicting the type of reference sequence used (c = coding DNA sequence, g = genomic reference sequence). When a protein-coding reference sequence is used (c), the nucleotide numbering begins with a 1, which represents the first position in the protein-coding region (the A of the translation-initiating ATG), and ends at the last position of the stop codon. Thus, if you divide the position number by 3, you can identify the affected amino acid in the protein sequence e.g., using the same example as above, 4375/3 = 1459, indicating that the predicted consequence affects amino acid 1459, which is an arginine. Different variants are indicated using different notations (explained in Table 2).
Table 2. HGVS notation and examples for the most common types of mutations2
Notation
Example
Explanation
>
c.4375C>T
Substitution of the C nucleotide at position c.4375 with a T
del
c.4375_4379del or c.4375_4379delCGATT
Nucleotides from position c.4375 to c.4379 deleted
dup
c.4375_4385dup or c.4375_4385dupCGATTATTCCA
Nucleotides from position c.4375 to c.4385 duplicated
ins
c.4375_4376insACCT
ACCT inserted between positions c.4375 and c.4376
delins
c.4375_4376delinsACTT or c.4375_4376delCGinsAGTT
Nucleotides from position c.4375 to c.4376 (CG) are deleted and replaced by ACTT
2.3. How to read mutation nomenclature: Predicted consequence
When only DNA has been analyzed, the RNA- and protein-level consequences of the variant can only be predicted, and should thus be reported in parentheses e.g., p.(Arg1459*) is the predicted effect at protein level (p) for the example described above.
3. The 3 prime rule for mutation nomenclature
For all variant descriptions using HGVS nomenclature, the nucleotide at the most 3’ position of the variation in the reference sequence is arbitrarily assigned to have changed (see how to apply this rule in Figure 2).4 The exception is for deletions/duplications around exon junctions for which shifting the variant 3’ would place it in the next exon.5
Figure 2. Application of the 3’ rule using the HGVS nomenclature recommendations for sequence variants.
4. Final thoughts and helpful tool
Although the HGVS recommendations can be difficult to understand and might take a bit of getting used to, if you break them down and refer to the examples in this guide, you are on the road to success!
If you want to accelerate your variant annotation and interpretation, Alamut™ Visual Plus is a comprehensive, full genome browser for efficient and user-friendly variant interpretation. The software accelerates the complex and time-consuming assessment of variants thanks to its user-friendly interface and integrated features for variant annotation and analysis.
Find out how Alamut™ Visual Plus applies the HGVS nomenclature recommendations to ensure that variant annotation follows the universally applied standards for variant analysis, interpretation, and reporting in our dedicated Technical Note.
Alamut™️ Visual Plus is for Research Use Only. Not for use in diagnostic procedures.
Whole exome sequencing (WES) is one of the latest advancements in next-generation sequencing (NGS) technology.This method sequences about 233,785 exons, or protein coding regions of the genome. This region is about 20,000 genes which makes up only about 1 percent of the human genome. Mutations in these regions can alter the respective proteins leading to various phenotypic implications. Analyzing the whole exome can unravel causative variants for diseases ranging from Mendelian to complex phenotypes.
When to use whole exome sequencing versus targeted or whole-genome sequencing
Targeted or panel sequencing only sequences genes that are known to be associated with a disease. It is often used, if the given disease has a clinical testing panel available and if the focus is solving the case with a short turnaround time.
However, if disease mechanisms are poorly understood, it can take several gene panels to identify the putative variant. Thus, WES allows for all protein-coding genes to be analyzed at once, which increases the probability of identifying the causative variant in a single sequencing assay. Since WES is not restricted to evaluating genes that have previously been associated with a specific disease, it gives a more comprehensive overview of the exome. Additionally, WES can help identify novel disease-gene associations in a research setting.
Another type of NGS is whole-genome sequencing (WGS), which evaluates the entire genome. This method can also be valuable for diseases with complex phenotypes or for cases where large structural variations are the primary cause of the disease. WGS can identify large structural variations and splicing variants in deep intronic regions. However, the large volumes of sequencing data pose some unique challenges for data analysis and storage. Although genomic sequencing continues to become cheaper and more accessible, it can be cost restrictive for some institutions.
Hence, WES can present a Goldilocks option between targeted gene panels and WGS. It generates a more comprehensive genomic view than targeted panels, but creates a data volume that is more manageable to analyze than that generated by WGS. In some specific cases, it also gives the researcher an opportunity to analyze copy number variations (CNV). Lastly, WES has a higher throughput and faster turnaround time compared to WGS.
Specific patient types who could benefit from whole exome sequencing
WES is an effective tool that can be used in a multitude of situations, but there are a few specific situations in which it presents significant advantages. One such situation is when an individual has overlapping phenotypes across multiple rare diseases. Using WES and taking a genotype-first approach can identify causal disease-gene associations and help diagnose overlapping conditions. This more comprehensive view can also shorten turnaround time for diagnosis compared with running a series of targeted panels.
The shorter turnaround time and higher diagnostic yield with WES also presents significant advantages for neonatal patients. Phenotypes can be difficult to assess in neonatal patients and some symptoms might not yet manifest. Comprehensive genetic testing such as WES can help to reach an early diagnosis, which not only allows for the implementation of an optimized care plan but can also provide information on any preventative measures and/or other potential complications that could arise as a result of the disorder.
Rare disease diagnosis is often an exhaustive journey for patients and family members seeking answers for their accurate diagnosis and disease management through several specialists, while undergoing multitude of tests and procedures, in pursuit of receiving the most effective treatment. WES can be a good follow-up to those initial tests, providing information that could uncover relevant variants in genes that were not previously implicated in a given disease (diagnosis).
Thus, WES cannot only be used for identifying causative variants related to the patient’s disorder, but the comprehensive approach can provide information about additional diseases or any genetic predispositions to other inherited disorders. This information is sometimes known as secondary or incidental findings and allows for patients to better manage their condition and understand the associated health risks. In some cases, this information may not be reported back to the patient, however, having the sequenced exomes allows for easier reanalysis of the data and future germline assessment of the same patient.
Considerations for implementing whole exome sequencing
A major logistical consideration for implementing WES beyond (the cost of) validating a new test is choosing which bioinformatics platform will be used to analyze the data. As touched on previously, WES produces much more data than sequencing using a targeted panel. Most whole-exome solutions sequence around 20,000 genes and a targeted panel that may sequence less than 100 genes. This larger volume of data is responsible for the advantages of WES, by creating a more comprehensive view of the genome which can increase diagnostic yield. Institutions that are interested in bringing on WES need to consider the large volume of data being generated and how this bioinformatic workflow can be challenging for finding relevant variants.
A bioinformatics platform for WES analysis should accurately and reliably identify multiple variant types in a single workflow and have a fast turnaround time and multiple filtering options to help streamline variant interpretation. The SOPHiA DDM™ Platform delivers advanced analytical performance, can complete WES analysis overnight, and has dedicated filtering features and a rich knowledgebase to help identify variants of interest associated with rare diseases for research purposes.
SOPHiA GENETICS products are for Research Use Only and not for use in diagnostic procedures unless specified otherwise.
SOPHiA DDM™ Dx Hereditary Cancer Solution, SOPHiA DDM™ Dx RNAtarget Oncology Solution and SOPHiA DDM™ Dx Homologous Recombination Deficiency Solution are available as CE-IVD products for In Vitro Diagnostic Use in the European Economic Area (EEA), the United Kingdom and Switzerland. SOPHiA DDM™ Dx Myeloid Solution and SOPHiA DDM™ Dx Solid Tumor Solution are available as CE-IVD products for In Vitro Diagnostic Use in the EEA, the United Kingdom, Switzerland, and Israel. Information about products that may or may not be available in different countries and if applicable, may or may not have received approval or market clearance by a governmental regulatory body for different indications for use. Please contact us to obtain the appropriate product information for your country of residence.
All third-party trademarks listed by SOPHiA GENETICS remain the property of their respective owners. Unless specifically identified as such, SOPHiA GENETICS’ use of third-party trademarks does not indicate any relationship, sponsorship, or endorsement between SOPHiA GENETICS and the owners of these trademarks. Any references by SOPHiA GENETICS to third-party trademarks is to identify the corresponding third-party goods and/or services and shall be considered nominative fair use under the trademark law.