Menu
Companion diagnostics (CDx) are medical devices, specifically an in vitro diagnostic device (IVD), providing important information regarding the safe and effective use of therapeutics. The Food and Drug Administration (FDA) ascribes them three crucial functions: 1) identify patients more likely to benefit from a therapeutic; 2) determine patients at increased risk of serious side effects; and 3) monitor treatment responses for the purpose of adjusting dosage or regimens to improve safety and effectiveness. In our estimation, CDx act as a compass that directs the healthcare provider to the most appropriate treatment for each patient1.
The inception of CDx can be traced back to 1998, when the FDA granted concurrent approval for trastuzumab, a targeted cancer drug, and HercepTest™, a HER2 immunohistochemical assay. This milestone marked the birth of the drug-diagnostic co-development model, a transformative approach that has since witnessed consistent and substantial adoption2.
However, over the next 14 years, CDx advancement was slow, with the majority of new approvals occurring only in the past decade. In fact, from 1998 to 2012, approximately 20 new CDx were approved, whereas from 2013 to 2023, that number rose to 1343.
Today, approximately 50% of all new molecular entity (NME) approvals in oncology have an associated CDx or biomarker listed in the label required for safe and effective use (based on FDA approvals from 2021 and 2022 of NME in oncology3). Despite the historical tendency toward oncology products, applications in rare diseases and metabolic syndromes are evolving, paving the way for CDx to become an intrinsic part of precision medicine clinical trials across many indications.
The use of a companion diagnostics strategy in clinical trials, which we define here as using one or more biomarkers to pre-select and enroll patients more likely to respond to the experimental therapy, is commonly employed in oncology. In these trials, identification and pre-selection have significant advantages, allowing smaller patient groups to power the statistical analysis, potentially reducing overall costs, and increasing the likelihood of approval4.
But while a CDx strategy makes regulatory approval of a cancer drug more likely, it can simultaneously add complexity to the process:
Despite the evident complexity, the widespread adoption of next-generation sequencing (NGS) has made using companion diagnostics and deploying biomarker-driven strategies in clinical trials easier by permiting screening for multiple biomarkers simultaneously. Rather than rely on a one-biomarker-one-test model, NGS permits patients to be screened for eligibility for multiple therapeutics or clinical trials.. Moreover, targeted gene panels and broader approaches, such as comprehensive genomic profiling (CGP) and whole exome sequencing (WES) have further streamlined the development of multi-biomarker-driven CDx.
Returning to our analogy, while CDx acts as the compass, NGS technologies are the roadmap to determine the most suitable treatment for each patient.
There are two well-defined pathways to approval for companion diagnostics:
HercepTest™ followed the first pathway where the physical kit, produced by an IVD manufacturer, received a Pre-market Approval (PMA). In contrast, the precedent for the single-site model was only established much later with the publication of clinical evidence supporting the hypothesis that BRCA-mutated patients were more likely to benefit from treatment with olaparib—a PARP inhibitor first approved for the treatment of advanced ovarian cancer5,6.
Requiring a complex workflow and expert oversight, BRCA analysis was more conducive to the simplicity of the single-site model since the very nature of this pathway streamlines validation – validating a workflow within a single lab is less time-consuming than across multiple labs. This new approach played a pivotal role in rapidly advancing the commercial adoption of NGS applications.
Today, most NGS-powered CDx assays follow the single-site pathway3, creating a new set of challenges. Despite the increased simplicity, this model confines assays to single locations, limiting the capacity for sample analysis, significantly increasing turnaround times, and reducing patient access. In the new age of precision medicine, these limitations are being addressed through a decentralized testing and analysis model supported by technology-agnostic and easy-to-implement workflows.
While direct co-development of a CDx and therapeutic stands as the preferred regulatory model by the FDA, alternative approaches may be utilized due to the inherent challenges of aligning IVD and drug development.
While the development of a CDx and therapeutic are tightly entwined, drug developers and IVD manufacturers remain separate entities with a few exceptions. This requires the carefully selection of partner(s) within the IVD and CDx ecosystem to ensure successful programs.
Many questions must be addressed early in the process, as even suboptimal approaches can significantly delay and impact commercial uptake:
The advent of NGS technologies has heralded a healthcare revolution, propelling us toward data-driven precision medicine. Yet, as we push ahead in developing biomarker-driven applications for a plethora of indications, we face the potential for increased implementation challenges, threatening to complicate the patient journey.
The adoption of a decentralized, globally accessible, intuitive, and technology-agnostic SOPHiA DDM™ Platform, leveraging proprietary algorithms and a vast portfolio of robust NGS-based applications, is well positioned to streamline CDx co-development and implementation, enhancing access to analytically robust solutions without overtaxing healthcare resources.
Our holistic approach to co-development is poised to chart a course toward a more integrated future, arming developers with the necessary data and insights to tackle potential hurdles and maximize the time and resources allocated to clinical research programs.
At SOPHiA GENETICS, our unwavering commitment is to collaboratively engineer deployable solutions that elevate implementation and accessibility in precision testing while streamlining the process of analysis and interpretation. Explore the possibilities of SOPHiA DDM™ for BioPharma by visiting our dedicated page.
References
1 Food and Drug Administration (FDA). In Vitro Companion Diagnostic Devices: Guidance for Industry and Food and Drug Administration Staff. Issued on: August, 2014. Accessed on: October, 2023. Retrieved from: https://www.fda.gov/media/81309/download
2 Jørgensen JT. Drug-diagnostics co-development in oncology. Front Oncol. 2014;4:208. doi: 10.3389/fonc.2014.00208
3 FDA. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). Accessed on: October 2023. Retrieved from: https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools
4 Biotechnology Industry Organization (BIO). Clinical Development Success Rates and Contributing Factors 2011–2020. Accessed on: October 2023. Retrieved from: https://go.bio.org/rs/490-EHZ-999/images/ClinicalDevelopmentSuccessRates2011_2020.pdf
5 Ledermann J, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852-61. doi: 10.1016/S1470-2045(14)70228-1.
6 Deeks ED. Olaparib: first global approval. Drugs. 2015;75(2):231-240. doi: 10.1007/s40265-015-0345-6
7 Hanna TP, et al. Mortality due to cancer treatment delay: systematic review and meta-analysis. BMJ. 2020 Nov 4;371:m4087. doi: 10.1136/bmj.m4087
Precision medicine, also known as personalized medicine, aims to enhance healthcare quality by tailoring treatments to each person's unique genetic makeup, environment, and lifestyle. While a fully individualized approach to medicine is still a work in progress, the recognition that patient heterogeneity influences treatment effectiveness is not new1.
Historically, medicine has heavily relied on trial-and-error strategies for discovering, developing, and testing new treatments targeted at specific indications. This disease-centered approach resulted in predetermined standard therapies tailored to the “average patient.” While this one-size-fits-all approach has succeeded in many indications, it also carries significant drawbacks, particularly when dealing with complex diseases such as cancer or inherited disorders. In these cases, the risk of adverse side effects (e.g., toxicity) and reduced therapeutic response often result in poorer patient prognoses and quality of life1.
Precision medicine represents a patient-centric paradigm shift, acknowledging each individual's uniqueness while using real-world data and advanced statistics to guide the discovery-to-treatment process. For instance, pharmacogenomics requires us to look at each patient genomic data individually and in the context of others, enabling stratification into cohorts for predicting treatment responses2.
Success in precision medicine hinges on the ability to derive meaningful insights from large patient datasets. Fostering data diversity has the potential to further advance progress in this area1,3.
Recent technological advances have made precision medicine more accessible and impactful than ever before. Next-generation sequencing (NGS) has become more affordable, transforming it from a research-focused technology into a tangible clinical reality. This progress was further propelled by the widespread adoption of electronic health records (EHRs) and laboratory information management systems (LIMS), which not only facilitate population-scale research but also enable the use of clinical decision support tools for the delivery of targeted therapies to individual patients4.
The ability to identify genetic biomarkers and assess variant pathogenicity has grown significantly in the past decade. This has not only revolutionized patient diagnosis but also transformed drug development. A pivotal moment was the approval of imatinib by the FDA in 2001, the first small molecule targeted therapy for chronic myeloid leukemia (CML)5. By inhibiting the BCR-ABL fusion protein, imatinib was shown to effectively slow the progression of CML from chronic phase to blast crisis, making it the first of its kind.
This groundbreaking milestone paved the way for the approval of many other targeted therapies, such as gefitinib targeting EGFR alterations associated with NSCLC (2003) and trastuzumab for HER2-positive breast cancer (2006). The pace of new targeted drug approvals continues to accelerate year after year, heralding a promising era of precision medicine6.
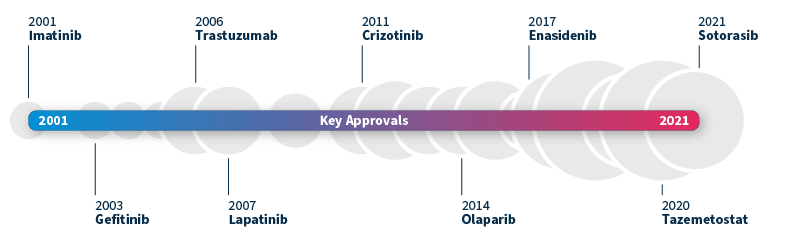
Timeline of FDA-approved targeted therapies in cancer. Grey bubbles represent the relative number of approvals per year. Data source: Waarts et al 2022.
To achieve a truly personalized approach to medicine, the harmonization of translational and precision medicine is paramount. This coordination between early mechanism-based drug development and late-stage patient-centric approaches gives rise to an end-to-end biomarker-guided process, allowing us to optimize treatment strategies for patient cohorts right from the outset7.
Known as translational precision medicine, this emerging concept brings a fresh perspective to the translational gap, calling for a broader scope beyond a purely genetic-based definition of biomarkers and introducing a multimodal approach by taking into account a wider range of healthcare variables. To make this new concept a reality, significant technological progress is required in several key areas8:
By addressing these critical areas of advancement, we can pave the way for a future where each patient receives personalized treatments tailored to her or his unique needs and characteristics. The pursuit of translational precision medicine promises to revolutionize healthcare, offering improved patient outcomes and transforming the landscape of medical research and development.
Powered by proprietary algorithms and enriched with data from 750+ institutions*, the SOPHiA DDM™ Platform accelerates advances in the field of precision medicine. Its core mission centers on empowering clinical researchers across healthcare and biopharma spheres alike.
To learn more about SOPHiA DDM™ BioPharma Solutions for biomarker-centric discovery, development, and application deployment, visit our page.
* The number of institutions represents active customers who have generated revenue through the SOPHiA DDM™ Platform usage or Alamut™ Visual Plus licenses as of September 30, 2022.
SOPHiA GENETICS products are for Research Use Only and not for use in diagnostic procedures unless specified otherwise.
SOPHiA DDM™ Dx Hereditary Cancer Solution, SOPHiA DDM™ Dx RNAtarget Oncology Solution and SOPHiA DDM™ Dx Homologous Recombination Deficiency Solution are available as CE-IVD products for In Vitro Diagnostic Use in the European Economic Area (EEA), the United Kingdom and Switzerland. SOPHiA DDM™ Dx Myeloid Solution and SOPHiA DDM™ Dx Solid Tumor Solution are available as CE-IVD products for In Vitro Diagnostic Use in the EEA, the United Kingdom, Switzerland, and Israel. Information about products that may or may not be available in different countries and if applicable, may or may not have received approval or market clearance by a governmental regulatory body for different indications for use. Please contact us to obtain the appropriate product information for your country of residence.
All third-party trademarks listed by SOPHiA GENETICS remain the property of their respective owners. Unless specifically identified as such, SOPHiA GENETICS’ use of third-party trademarks does not indicate any relationship, sponsorship, or endorsement between SOPHiA GENETICS and the owners of these trademarks. Any references by SOPHiA GENETICS to third-party trademarks is to identify the corresponding third-party goods and/or services and shall be considered nominative fair use under the trademark law.